The Mass Spectrometry and Proteomics core facility is available to all University of Nebraska campuses' investigators. Outside investigators are also welcome at a slightly higher rate.
MSPCF maintains and uses equipment for protein separation and imaging, as well as for sample preparation for mass spectrometry analysis. We are equipped with a wide range of bioinformatics tools for data mining and evaluation.
SPECIAL NOTE: If you are interested in submitting samples to the MSPCF, we strongly recommend that all persons involved in the project (PI, graduate students, post-doc, techs, etc.) schedule an initial consultation meeting with Dr. Vikas Kumar (Director), so we can assist you in choosing the right methods for your mass spectrometry experiment. This can save you time and money. In addition, we specialize in the development of customized projects outside of our routine analyses.
To submit samples for analysis, please complete the online submission form.
Samples can be dropped off at DRC1 1027 or 1050. Please email ahead to schedule a time for that.
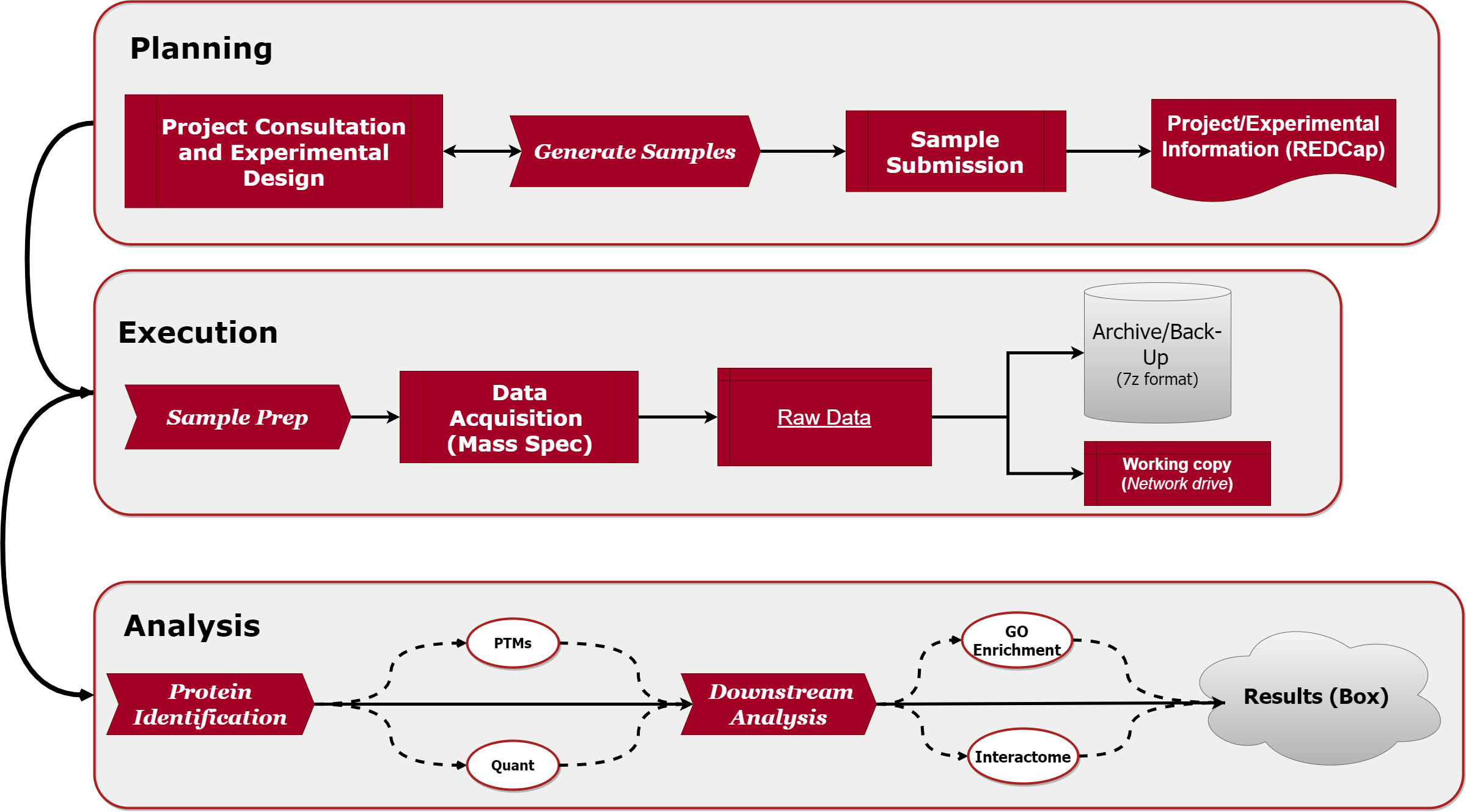
Our operations flow chart:
Services
Protein Identification – Protein IDs
This is the most common request by our users. This is accomplished by proteolytic digestion, followed by LC-MS/MS. We usually perform all steps of the protocol for you but will need to discuss the best method and approach for your particular project. After MS/MS data is collected, spectra are being correlated against a specific database, that you must specify in Sample submission form. After the analysis of spectra is completed, we will send out the list of all proteins detected in your samples, with all additional information, including a method section that can be used in publications.
Protein Interactome Analysis
If you are sending us a pooldown (IP), we suggest eluting proteins from the beads before sending it to us. This can be done using either high pH (0.5 M ammonium-hydroxide/0.5 mM EDTA, pH 11.0) or low pH (0.2 M glycine, pH 3.0). You may also decide to boil your sample in a sample buffer, but this is not our preferred method. Please neutralize the sample to around pH 7.5 and bring it to us on dry ice. We will start from there. As of the protocol details, it is similar to Protein Identification. For this type of experiment, you will have to have at least 3 biological replicates, and some sort of a negative control (usually IgG). You are also encouraged to confirm your specific protein with Western blot, before we start the prep for MS.
Protein Post-translational Modifications (PTMs)
Mass spectrometry has a capability to detect and characterize most fixed PTMs that are stable in proteins. The greatest challenge in PTM detection is their occurrence and frequency, as most of them are rare and dynamic events. PTM projects have to be well researched and discussed, and almost always involve specific biochemical enrichment strategies. For the determination of one, or multiple PTMs, it is better to start with a single highly purified protein in SDS-PAGE gel slice. This is our recommendation, rather than a rule. The protocol will involve a careful selection of multiple enzymes (2 or 3 enzyme approach) to maximize peptide coverage for specific sites of interest.
Quantitative MS
Determination of a change in protein expression between two or more samples has become extremely popular, but also particularly challenging. With the right approach and bioinformatics tools, we can analyze differences in protein expression in many studying systems, for example between healthy and sick animals or cancer cell lines, wild type and knockout organisms, cell lines with different treatments etc. For this, we have two approaches: labeled or label-free quantitative MS methods.
There are several very well-established methods that utilize labelling of proteins or peptides for their quantitative MS/MS analyses, and the one mostly used in our core is TMT labelling (tandem mass tags). These isobaric small chemical tags have the same structure and covalently attach to the free amino acid termini of Lys residues on a peptide. During MS/MS analysis, where peptides are being isolated and fragmented, each tag produces a unique reporter ion signature that makes quantitation possible. Protein quantitation is done by comparing the intensities of reporter ions.
Other very popular quantitation method is label-free MS/MS. The method has shown good results with highly reproducible chromatography step. Data is generated using either Maxquant, Proteome Discoverer or Scaffold.
MW determination
Using either MALDI or LC-based mass spectrometry, we can determine the high accuracy molecular weight of the specific biomolecule. Your sample should be without salts and detergents.
Rates
For assistance in experimental design of mass spectrometry and proteomics based projects or to inquire about establishing a research-based collaboration, contact the Drs. Kumar or Lagundzin.
| Service | Unit | Cost |
|---|---|---|
| Simple protein identification (60-min method) | Per sample | $70 |
| Complex protein identification (120-min method) | Per sample | $130 |
| Custom methods/Bulk submission (more than 100 samples) | Per sample | Inquire |
These services are done on Orbitrap Fusion Lumos (Thermo Sci) or 6600 TTOF (Sciex), and include 1 database search
| Service | Unit | Cost |
|---|---|---|
| Proteins and peptides <25 kDa (Autoflex MALDI, Bruker) | Per sample | $35 |
| Use of uncommon matrices (other than CHCA or SPA) | Per sample | $60 |
| Small molecules/metabolomics (QTRAP, Sciex) | Per hour | $70 |
| Service | Unit | Cost |
|---|---|---|
| Sample preparation | Per hour | $50 (cost of reagents NOT inculded) |
Examples of services available:
Protein and peptide quantification (colorimetric, fluorometric)
Peptide fractionation
In-gel digestion
In-solution digestion
Quantitative proteomics (labeled or label-free)
Sample clean-up
Sample concentration (centrifugal filters, vacuum-assisted)
Abundant protein depletion (albumin, IgG, …)
Phosphopeptide enrichment (IMAC, TiO2, …)
Coomassie Staining
Immunoprecipitation
Examples of additional costs:
Gels, trypsin, additional proteases, ZipTips, columns, filters, BCA, reagents for peptide quantification, plates...
It is recommended to ask for a quote for the more advanced services provided because sample prep times and cost of reagents can vary significantly: mspcf@unmc.edu
MSPCF is equipped with all necessery software and platforms for data analysis, please see more here: https://www.unmc.edu/vcr/cores/vcr-cores/mspcf/Bioinformatics.html
| Service | Unit | Cost |
|---|---|---|
| Additional database search | Per hour | $80 |
| Network/Functional enrichments analysis | Per hour | $85 |
| Consultations | Per hour | $85 |
| Training | Unit | Cost |
|---|---|---|
| Autoflex MALDI | Per hour | $80 |
| 6500 QTRAP | Per hour | $80 |
| Orbitrap Fusion Lumos | Per hour | Not available |
| 6600 TTOF | Per hour | Not available |
| Computer workstations | Unit | Cost |
|---|---|---|
| Proteome Discoverer (v 2.4) | Per hour | $5 |
| Protein Pilot | Per hour | $0 |
| MRM | Per hour | $0 |
| SWATH | Per hour | $0 |
| PDVC* | Per hour | $0 |
Workstations are located at DRC1 Room 1045. Reservations on RSS are required!
PDVC (Proteomics Data Viewer Computer) is shared computer for anyone who wishes to see their data using our software, such as Peaks, Scaffold etc. This workstation is located at DRC1 Room 1050. Please email us for inquiries: mspcf@unmc.edu
| Instrument use | Unit | Cost |
|---|---|---|
| Autoflex MALDI | Per hour | $40 |
| 6500 QTRAP | Per hour | $35 prime hours; $25 weekends and over-night runs 6 pm–8 am |
| 6600 TTOF* | Per hour | $50 |
* Time for blank runs between samples and after samples (mandatory) will be calculated in the total usage time
| Protein identification & PTM analysis | Unit | Cost |
|---|---|---|
| Simple protein identification (60-min method) | Per sample | $120 |
| Complex protein identification (120-min method) | Per sample | $240 |
| Custom methods/Bulk submission (more than 100 samples) | Per sample | Inquire |
| Molecular weight determination | Unit | Cost |
|---|---|---|
| Proteins and peptides <25 kDa (Autoflex MALDI, Bruker) | Per sample | $50 |
| Use of uncommon matrices (other than CHCA or SPA) | Per sample | $90 |
| Small molecules/metabolomics (QTRAP, Sciex) | Per hour | $120 |
| Service | Unit | Cost |
|---|---|---|
| Sample preparation | Per hour | $85 (cost of reagents NOT inculded) |
| Data analysis | Unit | Cost |
|---|---|---|
| Additional database search | Per hour | $120 |
| Network/Functional enrichments analysis | Per hour | $130 |
| Consultations | Per hour | $130 |
| Service | Instrument | Minimum amount* | Optimal amount |
|---|---|---|---|
| Protein identification | Orbirap Fusion Lumos | 15 fmol | 500 fmol |
| 6600 TTOF | 15 fmol | 500 fmol | |
| MALDI | 100 fmol | 500 fmol | |
| Molecular weight determination | MALDI | 100 fmol | 1 pmol |
* After tryptic digest and sample processing, expected recovery is 45-50%, but can be as low as 30%. This must be taken into account when submitting samples